
|
|
Jalview Documentation |
Input from the command line
When run as an application jalview takes the name of an alignment file on the command line. The format is :
java jalview.AlignFrame <alignfile> File|URL <format> [-mail <mailserver>-srsserver <srsserver> -database <srsdatabase>]For people using the script file jalview.bat or Jalview the syntax is
Jalview <alignfile> <format><alignfile> is the name of the alignment file which has to be in one of the supported formats. The allowed formats are MSF, FASTA, PIR, CLUSTAL and BLC.File|URL If you are reading from a local file use the File switch here. If you are reading a URL use the URL switch.
<format> This must be one of MSF, FASTA, PIR CLUSTAL, BLC, PFAM, MSP
Examples:
1) For a URL:
java jalview.AlignFrame http://circinus.ebi.ac.uk:6543/jalview/llym.msf URL MSF2) For a local fileJalview 1lym.blc BLCjava jalview.AlignFrame 1lym.pir File PIR<srsserver> The default is the EBI srs server :
srs.ebi.ac.uk/srs7bin/cgi-bin/ .If you use your own srs server then this option takes the location (minus the http://) of the wgetz program. In most cases this will beyour.hostname/srs7bin/cgi-bin/<database> The default database is swall which is probably EBI specific. Change this to your relevant database.See here for more details about SRS access.
Example for access to the sanger SRS site and the pfam database :
java jalview.AlignFrame myfile.pfam File PFAM -srsserver www.sanger.ac.uk/srs6bin/cgi-bin/ -database pfamseqInput from a URL
See Input from the command line for details of how to do this
Input from a local file (application only)
Selecting this option brings up a window where you can type in your local file and select the right format. Pressing the 'Apply' button loads up the alignment in a new window. Input formats allowed are MSF, FASTA, PIR, BLC, CLUSTAL, PFAM and MSP. Further details about formats are here.Output to a local file (application only)
This option allows you to save your alignment as text to a local file using much the same procedure as reading in a file. WARNING: files can be overwritten with no prompting.Outputting postscript to a local file (application only)
You can save your coloured alignment as postscript using this option. A window appears where you can select the font and fontsize you wish to use as well as whether the output orientation is portrait or landscape.Input via a text box
As applets can't write or read local files I have provided a way of inputting alignments by cutting and pasting.
Select your alignment from your local text editor or xterm and paste directly into jalview. You then have to select the format your alignment text is in and click apply to tell jalview to interpret the output. If the format is correct a new alignment window will appear.Unix selection: 'cat myalignfile' will display the alignment file on screen. Select the alignment text with the mouse. Move the mouse over the text input window and press the middle button. You should now have transferred your alignment to the jalview input box.
Windows selection: Open up your alignment in a text editor (notepad, wordpad or whatever). Select all the text with the mouse and type CTRL-C to copy it. Move your mouse over the jalview text input window and type CTRL-V. The alignment text should now be transferred to the input window.
Alignment output via a text box
Similarly to the textbox input option the text version of your alignment can be output via a java text box. See the 'Input via a text box' description for how to cut and paste your alignments.File formats supported
The formats supported are
- MSF (GCG output with no checksum),
- CLUSTAL (Clustalw output),
- FASTA (common and simple format),
- PIR (less common but almost as simple format) ,
- BLC (AMPS output)
- PFAM (simple and has the advantage of including start-end points).
Applet parameters
If you are running jalview via the button applet provided in the distribution then there are a number of parameters you can set to define different sequence groups and colour schemes. These are described in a separate document.
Selected sequences are used in the Colour and Calculate menus. The Consensus option in the Calculate menu only uses the selected sequences in its calculation and will display an error in its status bar if none are selected. Selecting a colour scheme in the Colour menu will only apply that scheme if any sequences are selected. If none are selected that colour scheme is applied to all sequences.
Sequences can also be selected in other displays such as the tree display window and the PCA results window. If a sequence is selected in one window it will also be selected in all the other windows.
All columns may be deselected by choosing the 'Deselect all columns'
option from the Edit menu.
To generate a new group or delete a group the buttons in the bottom right hand of the window can be used.
Tip: To generate a new group quickly :
To create a group from selected sequences or to see the available
sequence groups refere to the previous entry. By default when
jalview is first started all sequences are in the same group.
If Fastdraw is switched off other proportional fonts can be used
(Helvetica and Times) and the residues appear more spaced out on the screen.
The screen update time will also be slower (typically by a factor of 3).
Smaller font sizes (probably < 6) are of most use if the
text is switched off and the coloured residue boxes only are displayed
(see view menu).
If the redraw speed is too slow for you on your system then turning
off the boxes option and colouring the text black will speed it up
considerably.
| Residues | Description | Colour |
| ILVAM | Aliphatic/hydrophobic residues | pink |
| FWY | Aromatic | orange |
| KRH | Positive | red |
| DE | Negative | green |
| STNQ | Hydrophilic | mid blue |
| PG | Proline/Glycine (conformationally special) | magenta |
| C | Cysteine | yellow |
| Residues | Full name | Colour |
| V | Valine | |
| I | Isoleucine | |
| L | Leucine | |
| F | Phenylalanine | |
| Y | Tyrosine | |
| W | Tryptophan | |
| H | Histidine | |
| R | Arginine | |
| K | Lysine | |
| N | Asparagine | |
| Q | Glutamine | |
| E | Glutamate | |
| D | Aspartate | |
| S | Serine | |
| T | Threonine | |
| G | Glycine | |
| P | Proline | |
| C | Cysteine |
Choosing a colour (the colour selector)
Underneath the list of colours and residues is a panel where you can select the rgb values of any colour you wish to use. The user can either move the scrollbars to change the rgb values or type in the values (0-255) in the text boxes. The new colour will be displayed in the panel to the right of the scrollbars.
Changing colours (the residue panels)
Clicking on the colours assigned to different residues with the left mouse button will cause whichever colour is displayed in the colour selector to appear in that residue panel. If you wish to modify an existing residue panel colour right clicking that colour will change the the colour selector's colour to the residue panel colour. The colour selector colour can then be modified.
Changing residues
For each colour present in that scheme a list of residues it is applied to appears to its right. These residues can be moved or deleted or added to to group the residues in a different way. For instance you may just want to display the charged residues in one colour and the rest in another to highlight the charged ones or you may want to only colour the cysteines differently from the others.
If you wish to change the residues associated with a colour edit the residue string in the text field and press the 'Apply' button to its right. If any residues have been deleted from the text field they will be assigned a white colour and appear in the bottom residue panel. If any residues have been transferred from another colour panel they will be deleted from the old one. The main jalview alignment window will be automatically updated.
Any modifications of the colour scheme will only apply to sequences
that are selected in the main alignment window. This allows the user
to have multiple colour schemes in one alignment. If no sequences
are selected then the colour scheme applies to all sequences. BEWARE::
there is NO UNDO function.
This option depends on a consensus calculation having been performed. If no consensus exists (e.g. after a copy or a clustalw alignment) then no residues are coloured.
The PID option colours the residues (boxes and/or text) according to the percentage of the residues in each column that agree with the consensus sequence. Only the residues that agree with the consensus residue for each column are coloured.
| Percentage agreement | Colour |
| > 80 % | Mid blue |
| > 60 % | Light blue |
| > 40 % | Light grey |
| <= 40% | White |
When the features have finished transferring the features will be displayed
on the alignment with different colours for different features. The
colours are as follows
| Sequence feature | Colour |
| CHAIN | White |
| DOMAIN | White |
| TRANSMEM | Dark red |
| SIGNAL | Cyan |
| HELIX | Magenta |
| TURN | Green |
| SHEET | Yellow |
| STRAND | Yellow |
| CARBOHYD | Pink |
| ACT_SITE | Red |
| TRANSIT | Orange |
| VARIANT | Dark orange |
| BINDING | Blue |
| DISULFID | Dark yellow |
| anything else | Light gray |
When the features have been displayed on the alignment selecting a residue will change the display in the sequence feature console. The console will display details of any feature that has been selected and underneath a list of all features listed for that sequence.
There are at the moment a few limitations on the sequence feature
display:
>HBA_HUMAN/3-45
means the swissprot ID HBA_HUMAN starting at position 3 and ending
at position 45. If your alignment doesn't have the correct start end
positions the sequence feature overlay is at best
meaningless.
A good example of the usage of the start-end positions is the
Pfam database
of protein alignments.
If everything is configured correctly (srs server, database and alignment
ids) then you should get output like
the following :
![]()
The main window is coloured using all the features in the Pfam
pancreatic trypsin inhibitor alignment and the sequence feature console
shows details of all features at the selected residue (which is in between
2 disulphide bonds and at the active site in this case). In the background
can be seen the mini web browser showing the contents of a Swissprot entry.
When the editor first starts up the consensus sequence is automatically
calculated using all the sequences in the alignment and the PID colour
scheme is used as default. If the consensus option is selected again
only the currently selected sequences are used to calculate it and all sequences
in the alignment are coloured according to that consensus.
For each pair of sequences the best global alignment is found using BLOSUM62 as the scoring matrix. The scores reported are the raw scores. The sequences are aligned using a dynamic programming technique and using the following gap penalties :
Gap open :
12
Gap extend : 2
When you select the pairwise alignment option a new window will come up which will display the alignments in a text format as they are calculated. Also displayed is information about the alignment such as alignment score, length and percentage identity between the sequences.
If you want to save that pairwise alignment (it's not in any known
format I'm afraid) you can cut and paste it from the text window with the
mouse. You can also press the 'View in alignment editor' button
to bring up another editor window.
The version implemented here only looks at the clustering of whole sequences and not individual positions in the alignment to help identify functional residues. For large alignments plans are afoot to use the CORBA server written by Chris Dodge to do this 'residue space' PCA remotely.
When the Calculate->Principal component analysis option is selected all the sequences (not just the selected ones) are used in the calculation and for large numbers of sequences this could take quite a time. When the calculation is finished a new window is displayed showing the projections of the sequences along the 2nd, 3rd and 4th vectors giving a 3dimensional view of how the sequences cluster.
This 3d view can be rotated by holding the left mouse button down in the PCA window and moving it. The user can also zoom in and out by using the up and down arrow keys.
Individual points can be selected using the mouse and selected sequences show up green in the PCA window and the usual grey background/white text in the alignment and tree windows.
Different eigenvectors can be used to do the projection by changing
the selected dimensions in the 3 menus underneath the 3d window.
When the tree has been calculated a new window is displayed showing the tree with labels on the leaves showing the sequence ids. The user can select the ids with the mouse and the selected sequences will also be selected in the alignment window and the PCA window if that analysis has been calculated.
Selecting the 'show distances' checkbox will put branch lengths on the branches. These branch lengths are the percentage mismatch between two nodes.
Postscript output can be generated for this tree and mailed to you
by clicking the Output button. This will bring up a window asking
you for your email address and you can set font options and the page
orientation. Clicking the Apply button will generate the postscript
and send the email.
Selection and output options are the same as for the UPGMA tree.
Hierarchical analysis is based on each residue having certain physico-chemical
properties listed as follows:
In brief go about it like this :
![]() This link provides an example of the output after
grouping for Pfam family rnaseH:
This link provides an example of the output after
grouping for Pfam family rnaseH:
The grouping by tree may not be satisfactory and the user may want to edit the groups (Edit->Groups...) to put any outliers together.
Before selecting the conservation option change the colour scheme to something sensible (Taylor or hydrophobicity for example). When the conservation is done the existing colour scheme is modified so that the most conserved columns in each group have the most intense colours and the least conserved are the palest.
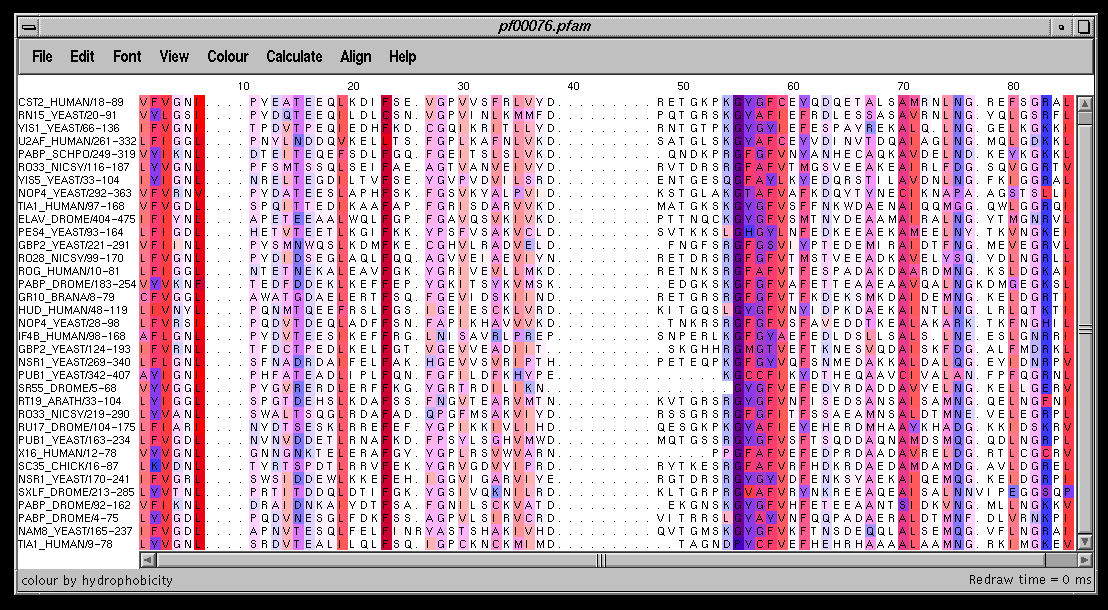
This link shows the results of first colouring the alignment by
hydrophobicity (Colour->by hydrophobicity) then performing conservation
analysis (Calculate->Conservation). Conserved hydrophobic columns
are shown with predominately red residues and conserved hydrophilic columns
with blue. The most conserved regions have the brightest colours.

Here is shown the same conservation but with Taylor colours instead
of hydrophobicity (Colour->Taylor).
The conservation analysis is done on each sequence group.
This highlights differences and similarities in conserved residue
properties between groups.
When this option is selected a window will appear giving you a message about whether your process is running and the time elapsed since the job was started. The cancel button will kill your process at any time.
The text box below should show the progress of your job but at the moment doesn't. I haven't been able to devise a simple way of displaying stdout as the alignment is progressing but I'm working on this. The stdout will appear in the xterm you started
When the alignment is finished a new alignment window is created with the aligned sequences in. No consensus calculation is done on these sequences by default so to see the similarity select Calculate->Consensus.
Due to applet security restrictions this option can only be used
from an application.
The Cancel button will cancel your job and the output is sent back to the text box below as the alignment progresses. As this application is written in 1.0 java (pretty much) to enable it to be used in older versions of netscape this display is somewhat flickery.
SRS server and database
The default SRS server and database are srs.ebi.ac.uk and swall at the EBI. To change to your own SRS server either use the -srsserver and -database options on the command line (see command line parameters) or use the
<param name="srsServer" value="srs.ebi.ac.uk/srs7bin/cgi-bin/">and
<param name="database" value="swall">options in the applet version.
Also for entries to be fetched correctly the sequence IDs in your alignment file must be of the right form.
The IDs must be :
HBA_HUMAN/6-20or just
HBA_HUMANwhere HBA_HUMAN is the sequence ID (not the accession) and the 6 and the 20 refer to the start and end residues of that sequence entry in the alignment. The start and end positions do not include gaps and are only essential if you wish to display the sequence features in the alignment window. I urge everyone to include these numbers as it stops embarrassing mistakes when inferring function from annotation of a multi-domain protein.
The application version now allows access to SRS through it's own mini web browser but at present none of the SRS links work (I'm trying to resist rewriting netscape :)
The right hand side of the status bar is mostly for development purposes but displays in milliseconds the time taken for the last redraw of the central sequence panel.